The Barthel Laboratory studies how genome instability shapes cancer, with a particular focus on telomere dysfunction and its consequences for the structure and evolution of brain tumors. We combine cytogenetics and molecular biology with long-read sequencing, 3D-genome assays, and computational genomics to resolve regions of the cancer genome that conventional pipelines miss.
Figure 1. Tumor phylogenetic trees (ancestry trees) linking multiple samples from three distinct gliomas projected into its original spatial context and onto a reference brain space. Samples from each tumor are shown by labeled points and the phylogeny (ancestry tree) was transformed in phylomorphospace using the sample coordinates as parameters. Lines represent phylogenetic (ancestral) relationships between samples. Tumor volumes are indicated by the gold (FLAIR) and orange-red (T1+Gado) overlays. Subcortical structures are shown as colored volumes. A reference brain volume (Montreal Neurological Institute) is shown in gray.
Research Projects
Telomere dysfunction and structural genome evolution
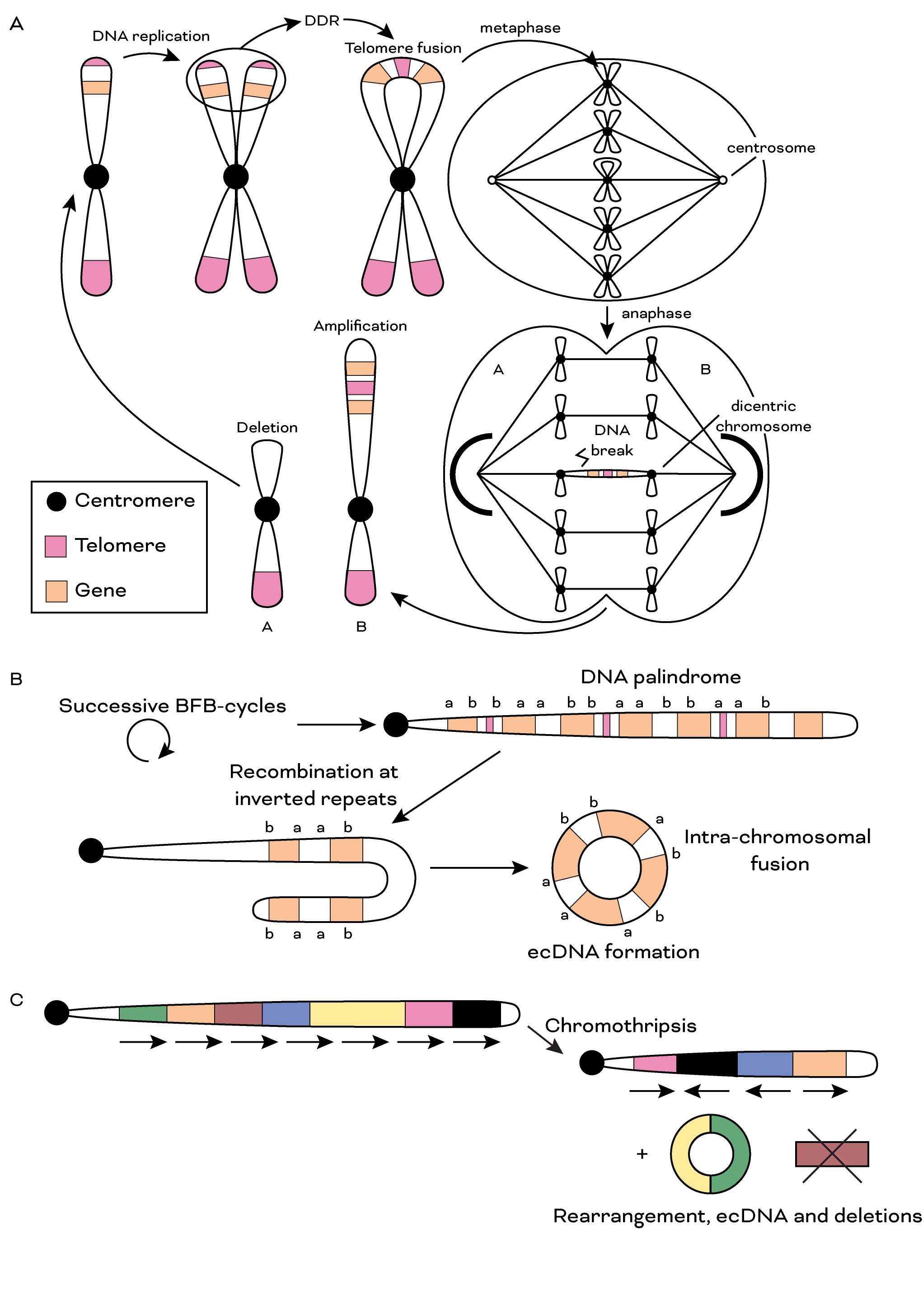
Cells that lose telomere protection acquire structural variants, including breakage-fusion-bridge cycles and chromothriptic rearrangements. Funded through the DOD CDMRP Career Development Scholar Award (HT9425-23-1-0844), we map these signatures in cell-based glioblastoma models, both through shelterin disruption (artificially induced telomere dysfunction) and gradual telomere erosion (spontaneous dysfunction), and are developing computational algorithms to detect equivalent patterns in patient tumor sequencing. Recent work shows that telomere dysfunction in normal human astrocytes (Mbegbu et al., 2026) drives acrocentric chromosome instability, with related findings on arm-specific telomere length in IDH-mutant astrocytoma (Jehangir et al., 2026).
Figure 2. Genomic instability related to telomere dysfunction. a. Schematic illustrating BFB-cycles. Following a single BFB-cycle, daughter cells are left with unequal DNA content, leading to unequal seggregation of genetic material. BFB-cycles may also involve fusion of non-sister telomeres (not shown). b. Successive BFB-cycles may form DNA palindromes demonstrating high intra-segmental homology. This can lead to intra-chromosomal fusions and formation of double minutes. c. Telomere dysfunction has also been linked to chromothripsis and following this phenomenon DNA segments can be rearranged, lost or circularized. DDR = DNA damage response; BFB = breakage-fusion-bridge.
Telomeres in 3D: chromatin structure around repetitive elements
Interphase telomeres make long-range chromatin contacts across the rest of the genome. We developed Telomere-C (Chen et al., 2025), a targeted Hi-C variant that enriches for telomeric contacts, and showed that the telomeric 3D interactome is anchored primarily at repetitive element hubs, including interstitial telomeric sequences (ITS), telomere-associated repeat 1 (TAR1), and D20S16 elements, with the D20S16 interaction uniquely enriched in cancer cells that use the alternative lengthening of telomeres (ALT) pathway. In a complementary line of work, telomere dysfunction in normal human astrocytes (Mbegbu et al., 2026) drives acrocentric chromosome instability and nucleolar reorganization, pointing to a mechanistic link between telomere maintenance and nucleolar structure.
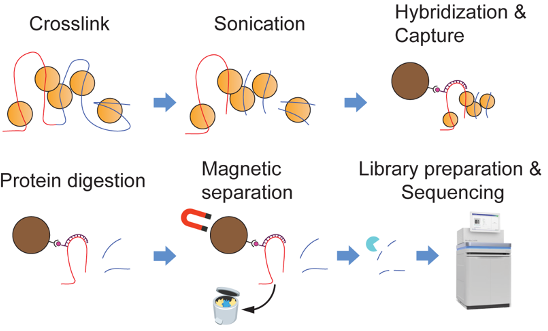
Figure 3. Telomere-C maps the telomeric 3D interactome. Telomere-C enriches for long-range chromatin contacts anchored at telomeres and reveals that the telomeric 3D interactome is dominated by ultra-long-range contacts with repetitive element hubs, including ITS, TAR1, and D20S16 elements.
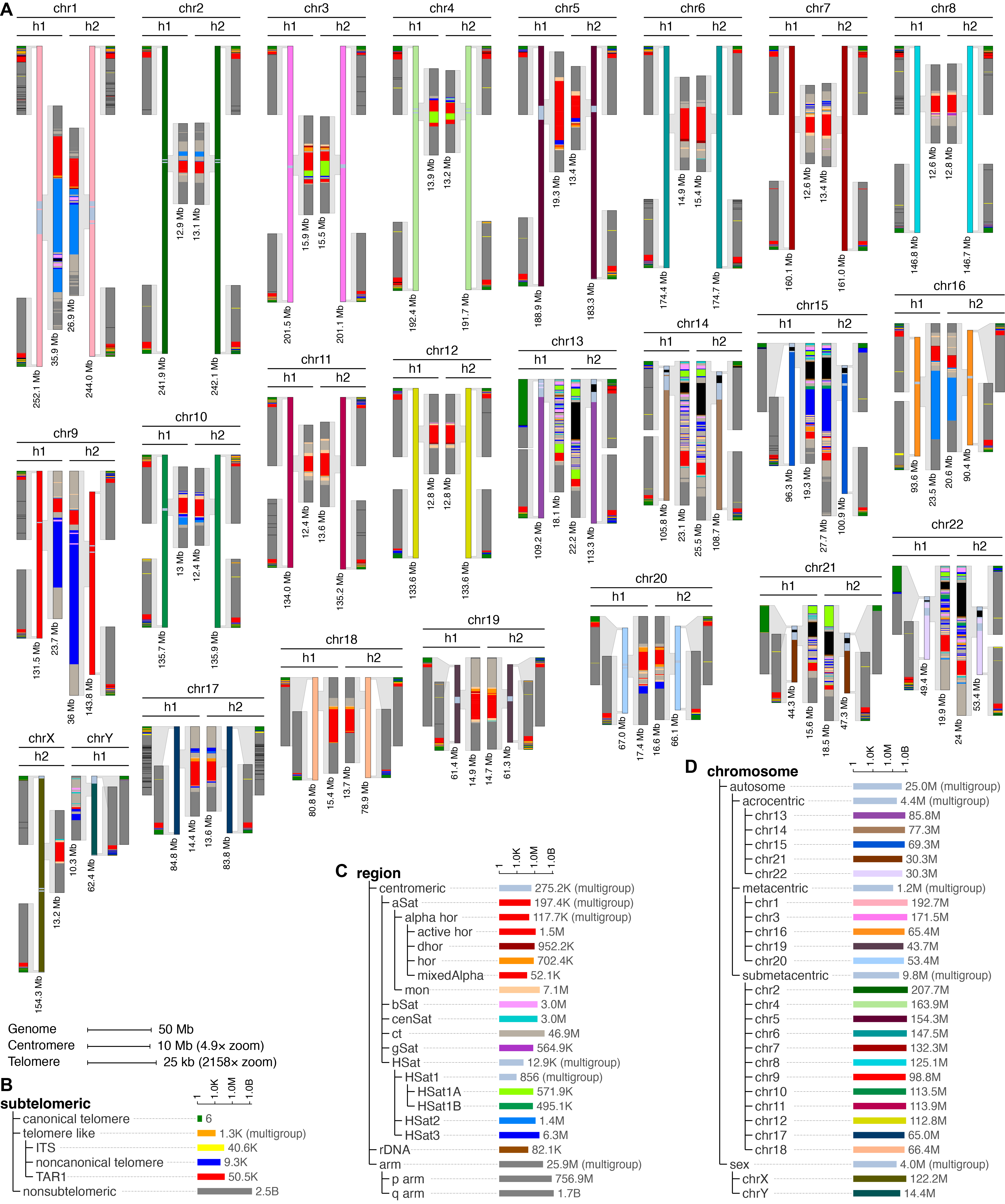
Computational genomics for the structurally complex genome
Centromeres, telomeres, and acrocentric short arms remain difficult to analyze with standard alignment-based pipelines, even on complete (T2T) assemblies. KaryoScope (Ranallo-Benavidez et al., 2026) is our alignment-free annotation tool that uses k-mer feature databases to generate karyotype-level annotations of human genomes in minutes. We applied it across the Human Pangenome Reference Consortium to catalog D4Z4 array diversity, centromere-scale variation, and Robertsonian translocation substrates; the same framework is used on diploid reference assemblies (BJ and IMR-90, NAR 2026) and cancer genomes (HG008, Wagner et al., 2026).
Figure 4. KaryoScope karyogram of HG002. Computational karyogram of a complete diploid human genome generated alignment-free by KaryoScope, with annotations for repeat families, satellite arrays, centromeric structures, and chromosome-end organization.
Liquid biopsies for longitudinal monitoring of cancer
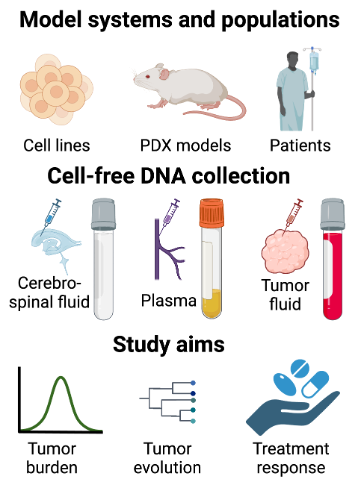
Tumors that are difficult to repeatedly sample, particularly glioma, benefit from cell-free DNA (cfDNA) monitoring of blood, cerebrospinal fluid, and other body fluids. Our cfDNA program combines shallow whole-genome sequencing with targeted panels, methylation profiling, fragmentomics, and mitochondrial-genome analysis to detect tumor-derived signal in CSF and plasma. Current applications include longitudinal CSF cfDNA monitoring of IL13Rα2 CAR-T therapy and PI3K-pathway resistance in glioblastoma (ABRC), and cfDNA monitoring within a CIRM-funded Phase I trial of neural stem cell-based oncolytic virotherapy. We also use in vitro host co-culture systems to evaluate cfDNA dynamics under chemotherapy (Mankame et al., 2025).
Figure 5. Cell-free DNA program across models and patient cohorts. Schematic of cfDNA collection (cerebrospinal fluid, plasma, tumor fluid) across cell lines, PDX models, and glioma patients, with study aims spanning tumor burden tracking, evolution monitoring, and treatment response assessment.
Multi-sample and post-mortem strategies for studying glioma evolution
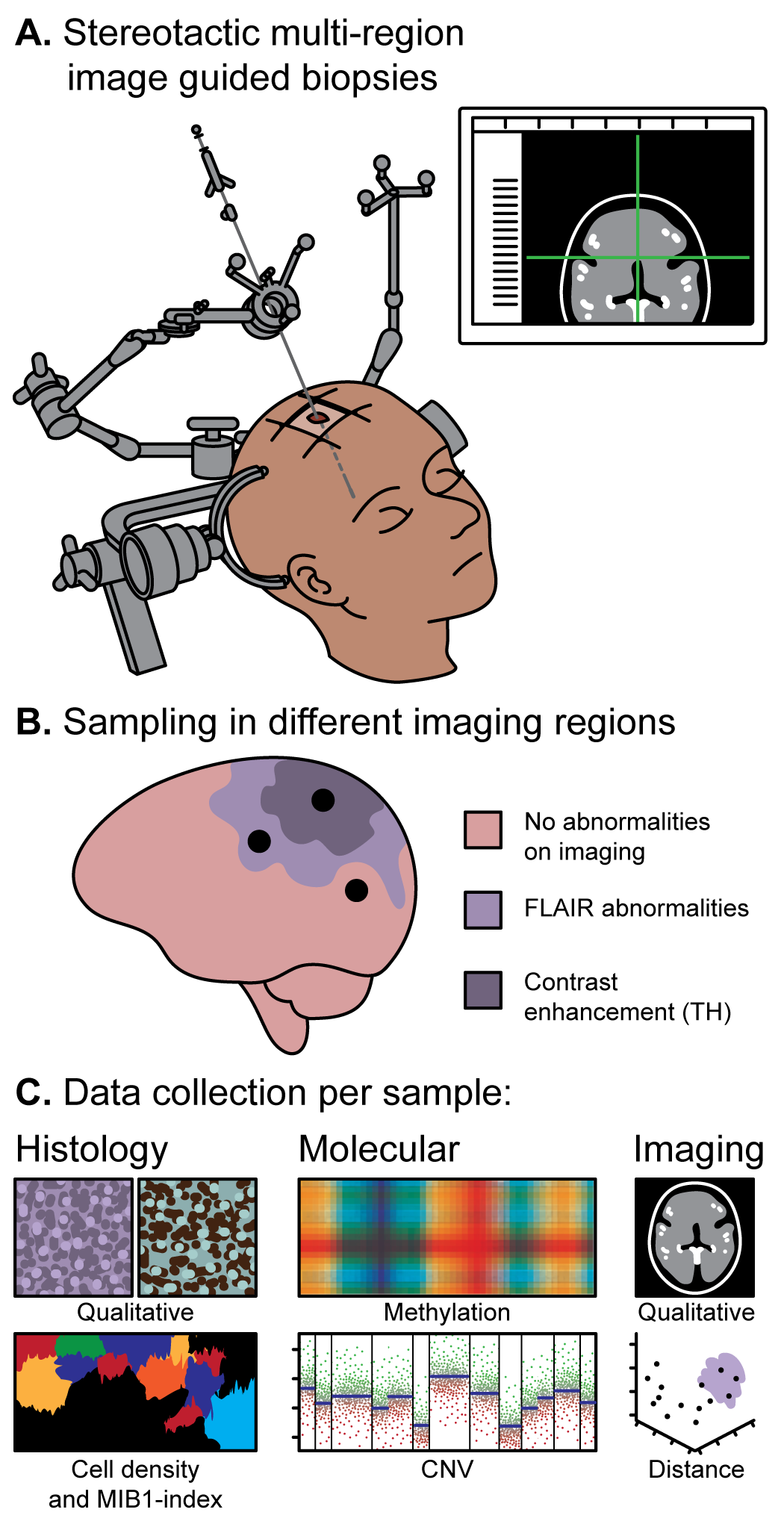
Most longitudinal glioma cohorts collect one or two samples per tumor, which limits what can be inferred about spatial-clonal architecture and treatment-driven evolution. Through the FRONTIER project, we collect MRI-mapped, pre-craniotomy multisector biopsies in IDH-wildtype and IDH-mutant glioma, paired with spatial transcriptomics and agent-based spatiotemporal modeling of glioma cell-state distributions (OPC-, APC-, and NPC-like); Verburg et al., 2021 is the methodological precedent for the sampling protocol. The LEGACY rapid-autopsy program is a complementary effort to collect post-mortem tissue from glioma patients at <6-hour post-mortem interval, providing matched terminal-disease specimens not represented in standard biobanks.
Figure 6. Multi-sector sampling across spatially distinct regions of a single tumor. Image-guided pre-craniotomy biopsies preserve MRI coordinates without brain shift, enabling reconstruction of spatial-clonal architecture across the tumor and adjacent infiltrated brain.