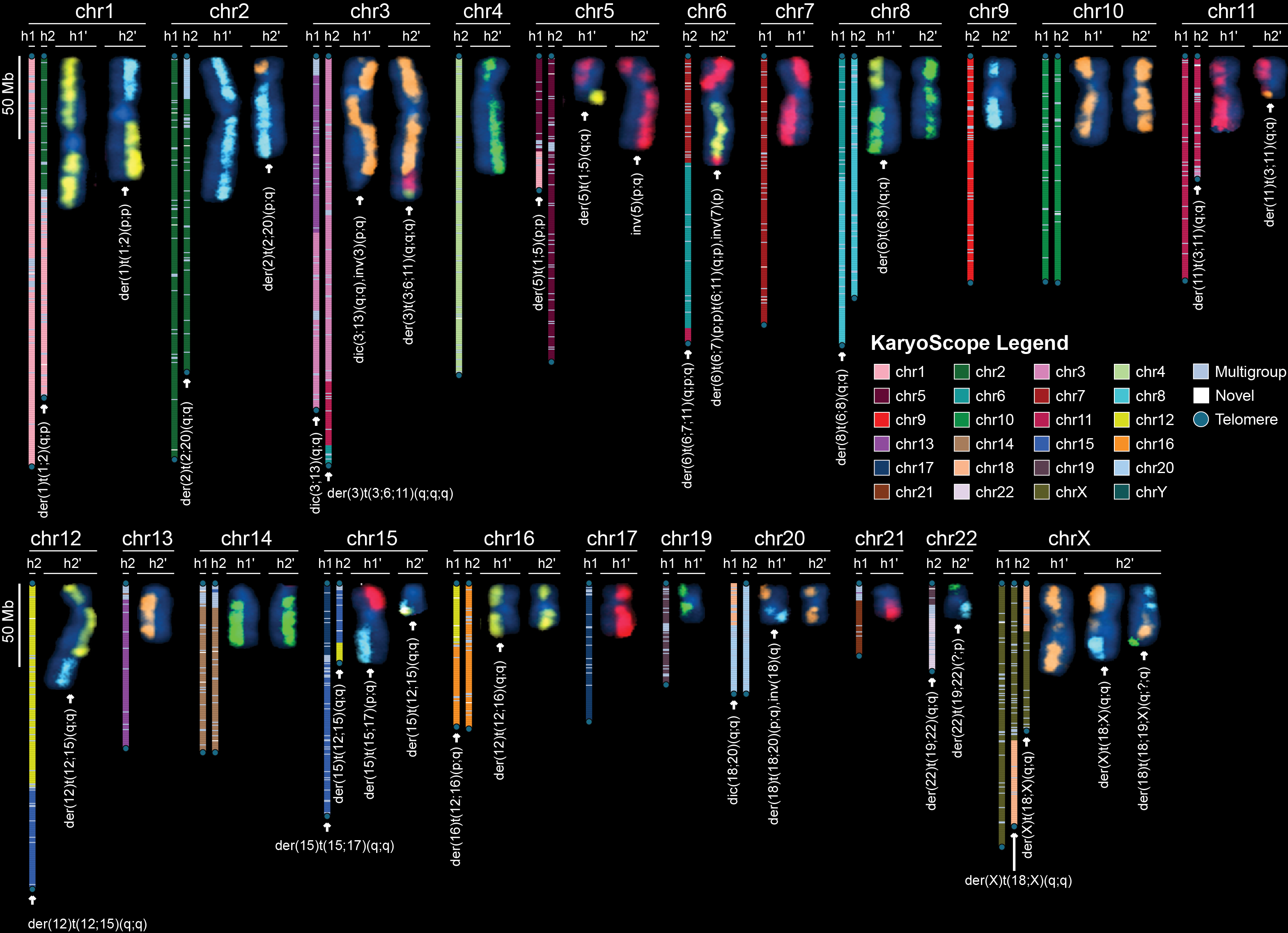
Cancer genome sequencing is essential for understanding tumor evolution and advancing precision medicine.1 However, reference gaps and germline variants obscure detection of small and large somatic variants and methylation in repetitive regions.1-3 It is common for tumor cells to gain or lose chromosome arms due to somatic structural changes that occur inside highly repetitive satellite DNA sequences in the centromeres.4 To identify the full spectrum of somatic variants, including complex rearrangements, we construct and curate near-complete, haplotype-resolved assemblies of the most recent common ancestor of an early-passage broadly-consented hypodiploid pancreatic cancer cell line and matched normal tissues. The tumor assembly completely recapitulates all 35 tumor chromosomes observed with karyotyping, with multiple translocation-induced hybrid chromosomes. The hybrid chromosomes contain putative functional dicentric and fused centromeres, nested foldback inversions causing 14 breakpoints with a haplotype switch in a single event, and centromeric satellite tandem duplications up to 136 kbp. Direct comparison of tumor and normal assembly haplotypes uncovers >7,000 variants altering >1 Mbp of sequence in repetitive regions that have been hidden by reference gaps and germline variants. 44 % of somatic small variants change representation because they alter germline variants on GRCh38, impacting mutational signatures and kataegis/omikli clusters. Most somatic LINE insertions originate from two hypomethylated non-reference germline LINE insertions, highlighting their impact on insertion mutation burden. These assemblies demonstrate that centromeric, acrocentric, and telomeric regions conventionally excluded from analysis harbor extensive somatic and epigenetic changes. Resolving complete tumor genomes enables a deeper understanding of cancer structural plasticity and the endpoints of breakage-fusion-bridge cycles. These assembled, curated paired normal-tumor benchmarks will serve as a critical foundation for developing future algorithms to characterize the most intractable regions of cancer genomes.
A complete human pancreatic cancer genome
Justin Wagner*, Ayse G. Keskus*, Keisuke K. Oshima*, T. Rhyker Ranallo-Benavidez*, Jennifer McDaniel, Mile Sikic, Dehui Lin, Luis F. Paulin, Adam C. English, Fritz J. Sedlazeck, Elizabeth M. Munding, J. Zachary Sanborn, Andrew Carroll, Pi-Chuan Chang, Daniel E. Cook, Kishwar Shafin, Joep de Ligt, Rayan Hassaine, Daniel Cameron, Severine Catreux, Yeonghun Lee, Lisa Murray, Sean Truong, Christian Brueffer, Aleksey V. Zimin, Erin Cross, Matthew McGowan, Michael Vernich, Andrew S. Liss, Jean-Pierre Kocher, Zachary Stephens, Tanveer Ahmad, Asher Bryant, Nathan Dwarshuis, Hua-Jun He, Zhiyong He, Nathan D. Olson, Francoise Thibaud-Nissen, Dmitry Antipov, Sergey Koren, Adam Phillippy, Rajeeva Lochan Musunuri, Giuseppe Narzisi, Miten Jain, Aaron M. Wenger, Stephen Eacker, Sayed Mohammad Ebrahim Sahraeian, Paul C. Boutros, Yash Patel, Takafumi N. Yamaguchi, Joseph McConnell, Matthew Borchers, Jennifer L. Gerton, Paxton Kostos, Andrea Guarracino, Maryam Jehangir, Hila Benjamin, Mohammed Faizal Eeman Mootor, Yuan Xu, Mobin Asri, Karen H. Miga, Jimin Park, Benedict Paten, Ruibang Luo, Zhenxian Zheng, Jae Young Choi, Linh Nguyen, Pankaj Vats, Dan R. Robinson, Josh N. Vo, Shenghan Gao, Ghulam Murtaza, Christopher E. Mason, Haoyu Cheng, Floris P. Barthel†, Chunlin Xiao†, Glennis A. Logsdon†, Mikhail Kolmogorov†, Justin M. Zook†
2026.
bioRxiv. 2026